Teo cơ tủy sống ( SMA ) là một chứng rối loạn thần kinh cơ hiếm gặp, đặc trưng bởi sự mất mát của các tế bào thần kinh vận động và sự xáo trộn cơ liên tục , thường dẫn đến tử vong sớm.
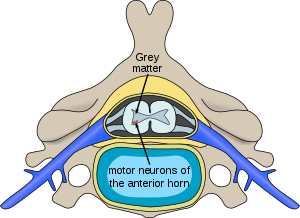
Rối loạn này được gây ra bởi một khiếm khuyết di truyền trong gen SMN1 , mã hóa SMN , một protein được biểu hiện rộng rãi trong tất cả các tế bào nhân chuẩn (tức là, các tế bào có nhân, kể cả tế bào người) và cần thiết cho sự tồn tại của các tế bào thần kinh vận động . Cấp thấp hơn của các kết quả protein đến mất chức năng của các tế bào thần kinh trong sừng trước của tủy sống và toàn hệ thống tiếp theo teo của cơ xương .
Teo cơ tủy sống biểu hiện ở mức độ khác nhau của mức độ nghiêm trọng, tất cả đều có sự suy giảm cơ bắp và suy giảm vận động cơ thường tiến triển. Cơ bắp gần , cơ tay và chân gần cơ thân và cơ hô hấp bị ảnh hưởng trước. Các hệ thống cơ thể khác cũng có thể bị ảnh hưởng, đặc biệt là ở các dạng khởi phát sớm của chứng rối loạn. SMA là nguyên nhân di truyền phổ biến nhất gây tử vong ở trẻ sơ sinh.
Teo cơ tủy sống là một rối loạn di truyền và được truyền theo một cách lặn tự phát. Vào tháng 12 năm 2016, nusinersen (được bán trên thị trường là Spinraza) trở thành thuốc được chấp thuận đầu tiên để điều trị SMA trong khi một số hợp chất khác vẫn còn trong các thử nghiệm lâm sàng.
Phân loại
SMA biểu hiện trên một loạt các mức độ nghiêm trọng, ảnh hưởng đến trẻ sơ sinh thông qua người lớn. Phổ bệnh được chia thành 3–5 loại, phù hợp với độ tuổi khởi phát triệu chứng hoặc với sự phát triển cao nhất của sự phát triển của cơ thể.
Phân loại được sử dụng phổ biến nhất như sau:
SMA1 (Trẻ con) :Còn gọi là Bệnh Werdnig – Hoffmann, Độ tuổi khởi đầu bình thường 0–6 tháng.
Đặc điểm : Dạng nặng biểu hiện trong những tháng đầu đời, thường là khởi phát nhanh và bất ngờ (“hội chứng em bé mềm”). Chết thần kinh vận động nhanh gây ra sự kém hiệu quả của các cơ quan chính của cơ thể – đặc biệt là hệ thống hô hấp – và suy hô hấp do viêm phổi là nguyên nhân tử vong thường gặp nhất. Trừ khi được đặt trên máy thở, trẻ sơ sinh được chẩn đoán mắc bệnh SMA loại 1 thường không sống qua hai tuổi, với tử vong xảy ra sớm nhất là trong vài tuần trong trường hợp nặng nhất (đôi khi được gọi là loại SMA 0 ). Với sự hỗ trợ hô hấp thích hợp, những người có kiểu hình SMA loại I nhẹ hơn, chiếm khoảng 10% trường hợp mắc bệnh SMA1, được biết là sống ở tuổi vị thành niên và trưởng thành.
SMA2 (Trung cấp) : Còn gọi là bệnh Dubowitz, độ tuổi khởi đầu từ 6–18 tháng.
Đặc điểm : Các hình thức trung gian ảnh hưởng đến trẻ em khiến chúng không bao giờ có thể đứng và đi bộ nhưng những người này có thể duy trì một vị trí ngồi ít nhất một số thời gian trong cuộc sống của họ. Sự khởi đầu của điểm yếu thường được nhận thấy một số thời gian từ 6 đến 18 tháng. Sự tiến bộ được biết là khác nhau rất nhiều, một số người dần dần phát triển yếu hơn theo thời gian trong khi những người khác thông qua điều trị cẩn thận tránh bất kỳ tiến triển. Vẹo cột sống có thể có mặt ở những trẻ này, và việc chỉnh sửa bằng nẹp có thể giúp cải thiện hô hấp. Cơ thể bị suy yếu, và hệ hô hấp là một mối quan tâm lớn. Tuổi thọ giảm nhưng hầu hết những người mắc bệnh SMA2 sống tốt khi trưởng thành.
SMA3 (Vị thành niên) : Còn gọi la Bệnh Kugelberg – Welander, độ tuổi khởi đầu > 12 tháng.
Đặc điểm : SMA loại 3 thường biểu hiện sau 12 tháng tuổi và mô tả những người mắc bệnh SMA3 có khả năng đi bộ mà không có sự hỗ trợ trong một thời gian, mặc dù nhiều người sau này đã mất khả năng này. Sự tham gia của hô hấp ít nhận thấy hơn và tuổi thọ bình thường hoặc gần như bình thường.
SMA4 (Người lớn khởi phát) : Khởi phát khi đã trưởng thành.
Đặc điểm : Dạng khởi phát của người trưởng thành (đôi khi được xếp loại là loại SMA khởi phát muộn) thường biểu hiện sau thập niên thứ ba (> 30 tuổi) của cuộc sống với sự suy yếu dần dần của cơ bắp – chủ yếu ảnh hưởng đến các cơ gần của tứ chi – thường yêu cầu người đó sử dụng xe lăn để di chuyển. Các biến chứng khác hiếm gặp và tuổi thọ không bị ảnh hưởng.
Dạng SMA nặng nhất đôi khi được gọi là SMA type 0 (hoặc SMA trẻ sơ sinh nặng) và được chẩn đoán ở trẻ sinh ra quá yếu đến nỗi chúng chỉ có thể tồn tại trong vài tuần ngay cả khi hỗ trợ hô hấp chuyên sâu. SMA type 0 không nên nhầm lẫn với SMARD1 có thể có các triệu chứng tương tự nhưng có nguyên nhân di truyền khác với SMA.
Phát triển vận động cơ thể ở những người mắc bệnh SMA thường được đánh giá bằng cách sử dụng thang đo chức năng đã được phê chuẩn – CHOP INTEND (Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders (Bệnh viện Nhi Philadelphia kiểm tra rối loạn thần kinh của trẻ em)) trong SMA1; và cả thang đo chức năng Hammersmith Functional Motor Scale trong vài biến thể trong loại SMA 2 và 3.
Dấu hiệu và triệu chứng
Các triệu chứng khác nhau tùy thuộc vào loại SMA, giai đoạn của bệnh cũng như các yếu tố riêng lẻ. Các dấu hiệu và triệu chứng dưới đây phổ biến nhất ở loại SMA nặng 0 / I:
- Không phản xạ , đặc biệt là ở chi dưới
- Tổng thể yếu cơ , giai điệu cơ yếu , khập khiễng hoặc xu hướng thất bại
- Khó đạt được các mốc phát triển, khó khăn khi ngồi / đứng / đi bộ
- Ở trẻ nhỏ: áp dụng vị trí chân ếch khi ngồi (hông bị bắt cóc và đầu gối uốn cong)
- Mất sức mạnh của các cơ hô hấp : ho yếu , khóc yếu (trẻ sơ sinh), tích tụ dịch tiết trong phổi hoặc cổ họng, suy hô hấp
- Thân hình chuông (gây ra bởi chỉ sử dụng cơ bụng để hô hấp) trong loại SMA nặng
- Sự co giật của lưỡi
- Khó bú hoặc nuốt, ăn kém
Nguyên nhân
Teo cơ tủy sống có liên quan đến đột biến di truyền trong gen SMN1 .
Nhiễm sắc thể người chứa hai gen gần như giống hệt nhau tại vị trí 5q13: một bản sao telomeric SMN1 và một bản sao trung tâm SMN2 . Ở những người khỏe mạnh, gen SMN1 mã hóa sự tồn tại của protein thần kinh vận động (SMN), như tên gọi của nó, đóng một vai trò quan trọng trong sự tồn tại của các tế bào thần kinh vận động . Mặt khác, gen SMN2 – do sự biến đổi trong một nucleotide đơn (840C → T) – trải qua nối thay thế tại điểm giao nhau của intron 6 đến exon 8, chỉ với 10-20% SMN2 bảng điểm mã hóa sự tồn tại đầy đủ chức năng của protein thần kinh vận động (SMN-fl) và 80-90% bảng điểm dẫn đến một hợp chất protein cắt ngắn (SMNΔ7) bị thoái hóa nhanh chóng trong tế bào.
Ở những người bị ảnh hưởng bởi SMA, gen SMN1 bị đột biến theo cách mà nó không thể mã hóa chính xác protein SMN – do việc xóa bỏ xảy ra ở exon 7 hoặc đột biến điểm khác (thường dẫn đến chuyển đổi chức năng của SMN1 trình tự vào SMN2 ). Tuy nhiên, hầu như tất cả mọi người đều có ít nhất một bản sao chức năng của gen SMN2 (với hầu hết là 2-4 trong số đó) vẫn mã hóa một lượng nhỏ protein SMN – khoảng 10-20% mức bình thường – cho phép một số tế bào thần kinh tồn tại . Về lâu dài, giảm sự sẵn có của các protein SMN trong cái chết dần dần của tế bào thần kinh vận động trong sừng trước của tủy sống và não. Cơ bắp phụ thuộc vào các nơron vận động cho đầu vào thần kinh innervation hiện nay đã giảm (còn gọi là denervation ), và do đó đã giảm đầu vào từ hệ thống thần kinh trung ương (CNS). Giảm truyền xung qua các tế bào thần kinh vận động dẫn đến giảm hoạt động co bóp của cơ bị đứt dây thần kinh. Do đó, cơ bắp bị từ chối trải qua teo tiến bộ (chất thải đi).
Cơ bắp của chi dưới thường bị ảnh hưởng đầu tiên, tiếp theo là cơ bắp của chi trên, cột sống và cổ và, trong trường hợp nghiêm trọng hơn, cơ phổi và co cứng. Cơ bắp gần như luôn luôn bị ảnh hưởng sớm hơn và đến một mức độ lớn hơn.
Mức độ nghiêm trọng của các triệu chứng SMA thường có liên quan rộng đến việc các gen SMN2 còn lại có thể bù đắp cho sự mất chức năng của SMN1 như thế nào . Điều này một phần liên quan đến số lượng các bản sao gen SMN2 có trên nhiễm sắc thể. Trong khi những người khỏe mạnh mang hai bản sao gen SMN2 , những người mắc bệnh SMA có thể có bất cứ thứ gì từ 1 đến 4 (hoặc nhiều hơn), với số lượng bản sao SMN2 càng lớn thì mức độ nghiêm trọng của bệnh càng nhẹ. Do đó, hầu hết các em bé mắc bệnh SMA loại I có một hoặc hai bản sao SMN2 ; những người mắc bệnh SMA II và III thường có ít nhất ba bản sao SMN2 ; và những người bị SMA IV thường có ít nhất bốn trong số chúng. Tuy nhiên, sự tương quan giữa mức độ nghiêm trọng của triệu chứng và số bản sao SMN2 không tuyệt đối và dường như có các yếu tố khác ảnh hưởng đến kiểu hình bệnh.
Teo cơ tủy sống được thừa hưởng trong một mô hình lặn tự phát , có nghĩa là gen khiếm khuyết nằm trên một thể nhiễm sắc thể điển hình. Hai bản sao của gen khiếm khuyết – một từ mỗi cha mẹ – được yêu cầu để kế thừa các rối loạn: các bậc cha mẹ có thể là nơi lưu trữ và không bị ảnh hưởng cá nhân. SMA dường như xuất hiện de novo (tức là, không có bất kỳ nguyên nhân di truyền nào) trong khoảng 2-4% các trường hợp.
Teo cơ tủy sống ảnh hưởng đến cá nhân của tất cả các nhóm dân tộc, không giống như các rối loạn lặn tự phát nổi tiếng khác, chẳng hạn như bệnh hồng cầu hình liềm và xơ nang , có sự khác biệt đáng kể về tỷ lệ xuất hiện giữa các nhóm dân tộc. Tỷ lệ mắc chung của SMA, của tất cả các loại và trên tất cả các nhóm dân tộc, nằm trong khoảng 1 trên 10.000 cá thể; tần số gen là khoảng 1: 100, do đó, khoảng một trong 50 người là tàu sân bay. Không có hậu quả gì về sức khỏe khi trở thành người chuyên chở. Một người chỉ có thể biết tình trạng của người vận chuyển nếu con của họ bị ảnh hưởng bởi SMA hoặc bằng cách giải mã gen SMN1 .
Các anh chị em bị ảnh hưởng thường có dạng SMA rất giống nhau. Tuy nhiên, sự xuất hiện của các loại SMA khác nhau giữa các anh chị em vẫn tồn tại – trong khi hiếm, những trường hợp này có thể là do thêm de novo xóa của SMN gen, không liên quan đến việc NAIP gen, hoặc sự khác biệt trong SMN2 sao chép số.
Chẩn đoán
Biểu hiện nghiêm trọng nhất trên phổ SMA có thể được chú ý đến các bà mẹ vào cuối thai kỳ do các chuyển động của thai nhi giảm hoặc vắng mặt. Các triệu chứng rất quan trọng (bao gồm suy hô hấp và ăn kém) thường dẫn đến tử vong trong vòng vài tuần. So với kiểu hình nhẹ nhất của SMA (người lớn khởi phát), nơi yếu cơ có thể xuất hiện sau nhiều thập kỷ và tiến tới việc sử dụng xe lăn nhưng tuổi thọ không thay đổi.
Các biểu hiện lâm sàng phổ biến hơn của phổ SMA nhắc chẩn đoán xét nghiệm di truyền:
- Suy nhược cơ song phương tiến triển (Thường là cánh tay và chân cao hơn so với bàn tay và bàn chân) trước một khoảng thời gian không triệu chứng (tất cả trừ loại nặng nhất 0)
- Làm phẳng thành ngực khi hít thở và nhô ra bụng khi hít vào.
- Hạ huyết áp kết hợp với không có phản xạ.
Trong khi các triệu chứng trên hướng tới SMA, chẩn đoán chỉ có thể được xác nhận với độ chắc chắn tuyệt đối thông qua xét nghiệm di truyền cho việc xóa bỏ al-7 của gen
SMN1 là nguyên nhân gây ra hơn 95% trường hợp. Xét nghiệm di truyền thường được thực hiện bằng cách sử dụng mẫu máu, và MLPA là một trong những kỹ thuật xét nghiệm di truyền thường được sử dụng nhiều hơn, vì nó cũng cho phép thiết lập số lượng bản sao gen SMN2.
Quản lý bệnh
Việc quản lý lâm sàng của một người mắc bệnh SMA thay đổi dựa trên mức độ nghiêm trọng / loại. Quản lý bệnh nhân có cùng loại SMA có thể thay đổi. Ở những dạng nặng nhất (loại 0/1), các cá nhân có điểm yếu cơ lớn nhất cần được can thiệp kịp thời. Trong khi tình trạng bệnh ít nghiêm trọng nhất (loại 4 / người lớn khởi phát), cá nhân có thể không tìm kiếm các khía cạnh chăm sóc nhất định cho đến sau này trong cuộc sống. Trong khi các loại SMA và cá nhân trong mỗi loại có thể khác nhau, do đó các khía cạnh cụ thể của việc chăm sóc cá nhân có thể khác nhau.
Chăm sóc hô hấp
Hệ hô hấp là hệ thống phổ biến nhất bị ảnh hưởng và các biến chứng là nguyên nhân gây tử vong hàng đầu ở các loại SMA 0/1 và 2. Loại SMA 3 có thể có các vấn đề hô hấp tương tự, nhưng hiếm gặp hơn. Các biến chứng phát sinh do cơ liên sườn suy yếu do thiếu sự kích thích từ dây thần kinh. Cơ hoành ít bị ảnh hưởng hơn các cơ liên sườn. Khi bị yếu đi, các cơ không bao giờ phục hồi hoàn toàn khả năng chức năng tương tự để giúp thở và ho cũng như các chức năng khác. Do đó, hơi thở khó khăn hơn và gây ra nguy cơ không nhận đủ oxy / hơi thở nông và không đủ thông khí tiết ra đường hô hấp. Những vấn đề này thường xảy ra khi ngủ, khi cơ bắp thoải mái hơn.
Cơ nuốt trong họng có thể bị ảnh hưởng, dẫn đến khó nuốt cùng với cơ chế ho khan làm tăng khả năng nhiễm trùng / viêm phổi . Hành động hỗ trợ bao gồm vật lý trị liệu ngực bằng tay hoặc cơ khí với hệ thống thiết bị hỗ trợ. Hỗ trợ thở, thông khí không phẫu thuật ( BiPAP)) thường được sử dụng. Phẫu thuật mở khí quản đôi khi có thể được thực hiện trong những trường hợp nặng hơn. Cả hai phương pháp thông khí kéo dài sự tồn tại đến một mức độ tương đương, mặc dù phẫu thuật mở khí quản ngăn chặn phát triển lời nói.
Dinh dưỡng
Loại SMA càng nghiêm trọng, càng có nhiều vấn đề về sức khỏe liên quan đến dinh dưỡng. Các vấn đề sức khỏe có thể bao gồm khó khăn trong việc cho ăn, mở hàm, nhai và nuốt. Những người có khó khăn như vậy có thể có nguy cơ gia tăng hoặc suy dinh dưỡng, không phát triển mạnh và khó nuốt, khó thở.
Các vấn đề về dinh dưỡng khác, đặc biệt là ở những cá nhân không được cấp cứu (các loại SMA nặng hơn), bao gồm thức ăn không đi qua dạ dày đủ nhanh, trào ngược dạ dày, táo bón, ói mửa và đầy hơi. Trong đó, có thể cần thiết trong loại SMA I và những người bị bệnh nặng hơn loại II có ống dẫn hoặc dạ dày . Ngoài ra, những bất thường về trao đổi chất do sự suy giảm β-oxy hóa của các axit béo trong cơ bắp và có thể dẫn đến tình trạng thiếu acid hữu cơ và tổn thương cơ bắp, đặc biệt là khi nhịn ăn.
Người ta cho rằng những người mắc bệnh SMA, đặc biệt là những người mắc bệnh nặng hơn, giảm lượng chất béo và tránh ăn chay kéo dài (ví dụ, ăn thường xuyên hơn người khỏe mạnh) cũng như chọn thực phẩm mềm hơn. Trong một bệnh cấp tính, đặc biệt là ở trẻ em, các vấn đề về dinh dưỡng có thể xuất hiện hoặc có thể làm trầm trọng thêm một vấn đề hiện tại (ví dụ: khó nuốt) cũng như gây ra các vấn đề sức khỏe khác như rối loạn điện giải và đường huyết.
Chỉnh hình
Các vấn đề về xương kết hợp với các cơ yếu trong SMA bao gồm các khớp bị co thắt với các chuyển động giới hạn, trật khớp hông, biến dạng cột sống, loãng xương, tăng nguy cơ gãy xương và đau. Các cơ yếu thường làm ổn định các khớp như cột sống dẫn đến sự phát triển của bệnh kyphosis và / hoặc vẹo cột sống và co bóp khớp. Sự hợp nhất cột sống đôi khi được thực hiện ở những người bị SMA I / II khi họ đạt đến 8-10 tuổi để giảm áp lực của cột sống bị biến dạng trên phổi. Hơn nữa, các cá nhân bất động, tư thế và vị trí trên các thiết bị di động cũng như các bài tập chuyển động và tăng cường xương có thể là quan trọng để ngăn ngừa các biến chứng. Những người mắc bệnh SMA cũng có thể hưởng lợi rất nhiều từ nhiều hình thức vật lý trị liệu , liệu pháp massage hỗ trợ.
Hỗ trợ di chuyển
Dụng cụ chỉnh hình có thể được sử dụng để hỗ trợ cơ thể và hỗ trợ đi bộ. Ví dụ, các dụng cụ chỉnh hình như AFO (orthining mắt cá chân) được sử dụng để ổn định bàn chân và hỗ trợ dáng đi, TLSO (chỉnh hình thắt lưng ngực) được sử dụng để ổn định thân. Các công nghệ hỗ trợ có thể giúp quản lý vận động và hoạt động hàng ngày và làm tăng đáng kể chất lượng cuộc sống.
Tim mạch
Mặc dù trái tim không phải là một vấn đề thường xuyên quan tâm, một liên kết giữa SMA và một số điều kiện tim đã được đề xuất.
Sức khỏe tâm thần
Trẻ em SMA không khác với dân số chung trong hành vi của chúng; phát triển nhận thức của họ có thể nhanh hơn một chút, và một số khía cạnh của trí thông minh của họ cao hơn mức trung bình. Mặc dù bị khuyết tật, những người bị ảnh hưởng bởi SMA báo cáo mức độ hài lòng cao từ cuộc sống.
Chăm sóc giảm nhẹ trong SMA đã được chuẩn hóa trong Tuyên bố đồng thuận về tiêu chuẩn chăm sóc trong teo cơ tủy sống đã được khuyến cáo sử dụng tiêu chuẩn trên toàn thế giới.
Thuốc
Nusinersen là thuốc duy nhất được chấp thuận để điều trị teo cơ cột sống. Nó được tiêm trực tiếp vào hệ thống thần kinh trung ương bằng cách sử dụng tiêm trong vỏ . Nó được sự chấp thuận của Ủy ban châu Âu trong thủ tục tập trung vào tháng năm 2017.
Tiên lượng
Trong điều trị thiếu thuốc, những người mắc bệnh SMA có xu hướng xấu đi theo thời gian. Gần đây, sự sống còn đã tăng lên ở bệnh nhân SMA nặng với hỗ trợ hô hấp và hỗ trợ dinh dưỡng tích cực và chủ động.
Đa số trẻ em được chẩn đoán mắc bệnh SMA type 0 các vấn đề hô hấp tái phát là nguyên nhân chính gây tử vong. Với sự chăm sóc thích hợp, các trường hợp mắc bệnh SMA nhẹ hơn (chiếm khoảng 10% trong tất cả các trường hợp mắc bệnh SMA1) sống đến tuổi trưởng thành. Sự tồn tại lâu dài trong loại SMA Tôi không được chứng minh đầy đủ; tuy nhiên, những tiến bộ gần đây trong hỗ trợ hô hấp dường như đã làm giảm tỷ lệ tử vong.
Trong bệnh SMA loại II, tiến trình của bệnh chậm hơn và tuổi thọ ít hơn so với dân số khỏe mạnh. Cái chết trước tuổi 20 là thường xuyên, mặc dù nhiều người mắc bệnh SMA sống có thể trở thành cha mẹ và ông bà. SMA type III có tuổi thọ bình thường hoặc gần bình thường nếu tuân theo các tiêu chuẩn chăm sóc. Loại IV, SMA khởi phát ở người lớn thường có nghĩa là chỉ suy giảm khả năng vận động và không ảnh hưởng đến tuổi thọ.
Trong tất cả các loại SMA, vật lý trị liệu đã được chứng minh là làm chậm sự tiến triển của bệnh.
Xem thêm : Thư gửi cha mẹ có con bị mắc bệnh teo cơ tủy sống
Bài viết tham khảo nguồn tin từ tài liệu mở : Wikipedia.org
Ngày : 10-09-2018 . Một số thông tin có thể đã thay đổi theo kết quả các nghiên cứu mới hơn sau ngày cập nhật gần nhất.